
Comparative chloroplast genomic and phylogenetic analysis of Aralia and related species
-
摘要:
分别对楤木属(Aralia)和羽叶参属(Pentapanax)各两个物种进行叶绿体基因组测序、组装和注释,并进行结构分析;结合NCBI下载的近缘类群叶绿体组序列,进行系统发育分析。结果显示,4个物种的叶绿体基因组均为环状四分体结构,长度为155 744 ~ 156 201 bp,GC含量为38.1%,均包含132个基因,其中蛋白质编码基因87个,rRNA基因8个,tRNA基因37个。边界分析发现IR(Inverted repeat)区均未发生收缩和扩张。SSR(Simple sequence repeat)序列数量在39 ~ 43个,多为单核苷酸和二核苷酸重复,位置多在非编码区。序列差异多出现在LSC(Large single copy)和SSC(Small single copy)区的非编码区。最大似然树揭示出两个高度支持的单系分支:第1分支包括羽叶参属、浓紫龙眼独活(Aralia atropurpurea Franch.)、食用土当归(Aralia cordata Thunb.)和东北土当归(Aralia continentalis Kitagawa);第2分支则均由楤木属物种组成。总体来说,楤木属和羽叶参属植物的叶绿体基因组序列比较保守;系统发育分析结果支持羽叶参属置入楤木属内部以及楤木属的各个种成单系。
Abstract:The chloroplast genomes of two species of Aralia and Pentapanax were sequenced, assembled, annotated, and structurally analyzed. Furthermore, combined with chloroplast genome sequences of related species downloaded from the NCBI database, phylogenetic analysis was conducted. The chloroplast genomes of the four species all exhibited a quadripartite circular structure with a length of 155 744 – 156 201 bp and GC content of 38.1%. Both genera contained 132 genes, including 87 protein-coding genes, eight ribosomal RNA (rRNA) genes, and 37 transfer RNA (tRNA) genes. Boundary analysis found that neither contraction nor expansion occurred in the inverted repeat regions. The number of simple sequence repeat (SSR) loci ranged from 39 to 43, most of which were mononucleotide and dinucleotide repeats, and mostly located in non-coding regions. Sequence differences mainly occurred in the non-coding regions of the large single copy (LSC) and small single copy (SSC) regions. The maximum-likelihood tree revealed two highly supported monophyletic branches: the first composed of Pentapanax, Aralia atropurpurea Franch., Aralia cordata Thunb., and Aralia continentalis Kitagawa and the second composed of other Aralia species. In general, the chloroplast genome sequences of Aralia and Pentapanax plants were relatively conserved. The phylogenetic results supported the placement of Pentapanax into the genus Aralia, with each Aralia species forming a monophyly.
-
Keywords:
- Aralia /
- Pentapanax /
- Chloroplast genome /
- Phylogeny
-
地黄(Rehmannia glutinosa Libosch.)为玄参科地黄属植物,多年生草本,以块根入药,为著名的“四大怀药”之一。地黄最早记载见《神农本草经》,被列为上品,迄今已有2000余年的应用历史。根据炮制方法的不同,地黄药材分为鲜地黄、生地黄和熟地黄[1]。2020版《中国药典》记载,鲜地黄具有清热生津、凉血、止血的功效,生地黄具有清热凉血、养阴生津的功效,熟地黄具有补血滋阴,益精填髓的功效[2]。地黄富含环烯醚萜类、苯乙醇苷类、紫罗兰酮类、三萜类、黄酮类和糖类等,对人体心脑血管、血液、中枢神经和免疫系统等均有显著作用[3]。
毛蕊花糖苷(Acteoside)是地黄中含量较高的苯乙醇苷类化合物[4],具有抗氧化、免疫调节、抗炎、保肝、抗肿瘤、增强记忆力等生物活性[5],是2010版、2015版《中国药典》规定的地黄药材质量控制的指标性成分之一。地黄毛蕊花糖苷的含量易受品种[6]、产地[7]、收获时期[5]、种植密度[8]和光照条件[9]等因素的影响,造成某些年份部分地黄药材的毛蕊花糖苷含量达不到《中国药典》规定的要求。地黄毛状根中也含有丰富的毛蕊花糖苷,本课题组前期研究表明,在毛状根诱导的特定时期添加水杨酸(SA)可显著促进毛蕊花糖苷的含量[10]。然而,地黄生长发育过程中叶面喷施SA对毛蕊花糖苷的含量是否有影响还未见报道。
本研究以大田栽培的地黄为材料,采用叶面喷施方法分析SA对地黄叶片和块根毛蕊花糖苷含量的影响,并利用转录组测序技术分析地黄块根中的基因表达特征,研究结果旨在为生产中应用外源激素提高毛蕊花糖苷的含量提供理论依据。
1. 材料与方法
1.1 实验材料
供试材料为地黄‘温85-5’,种植在河南省武陟县蔡庄村(35°2′51″N,113°18′34″E),经河南农业大学王丰青教授鉴定为Rehmannia glutinosa Libosch.。将浓度为100 μmol/L的SA水溶液均匀喷施在生长180 d处于膨大后期长势一致的地黄叶片上,以叶片完全湿润且无水滴落下为准。分别于处理后1、3和6 h进行取样,取样时选择位置相同的叶片。对照组喷施蒸馏水,与处理组同时取样。样品清洗干净后,将块根切成1 cm3左右的小块,叶片剪碎,55℃烘干后打粉,过3号药典筛。样品粉末存放于干净的8号自封袋中备用。
1.2 毛蕊花糖苷含量测定
对照品毛蕊花糖苷(批号MUST-18032725)购于成都曼斯特科技有限公司,纯度均 ≥ 98%。采用 Agilent1260高效液相色谱仪(美国安捷伦科技有限公司)进行检测。
色谱条件:采用的色谱柱型号为 Dikma Diamonsil C18(4.6 × 250 mm,5 μm),柱温30℃,流速1 min/mL。毛蕊花糖苷的流动相为乙腈−0.1%醋酸水(16 : 84),检测波长334 nm,进样量为20 μL。
供试样品溶液制备:精密称取地黄叶和块根样品粉末0.8 g,放入锥形瓶中,精密吸取50 mL甲醇加入锥形瓶中,称重,并在65℃加热回流提取1.5 h,放凉至室温称重,用甲醇补足失重后,摇匀过滤。于蒸发皿中精密吸取滤液20 mL进行毛蕊花糖苷分析,在电热恒温水浴锅上浓缩至近干,残渣用流动相溶解,转移至5 mL容量瓶中,用流动相稀释至刻度,摇晃均匀,用0.22 μm微孔的滤膜过滤,滤液装入2 mL的进样瓶待测。
含量计算:以本实验室建立的标准曲线Y = 30024X − 110.9来计算毛蕊花糖苷含量,Y为峰面积积分值,X为样品的质量浓度。
1.3 样品RNA提取及高通量测序
用TRIzol试剂提取样品的总RNA,用核酸测定仪检测RNA的浓度和质量。使用带有Oligo dT的磁珠富集具有polyA尾巴的mRNA,然后将RNA片段化,反转录后再合成cDNA第2链,形成双链cDNA。双链cDNA经过末端修复、3′末端加A、添加测序接头、多轮扩增、热变性成单链及单链环化等一系列步骤后,完成测序文库的制备。测序委托华大基因科技有限公司进行,测序平台为BGISEQ-500。
对测序得到的原始序列(Raw reads)进行质控处理,去除低质量、接头及污染序列,获取过滤后的测序序列(Clean reads)。使用Bowtie2将Clean reads比对到课题组前期获得的地黄叶和根的参考基因序列集[11],统计不同样品的片段序列比对率以及分布,之后再使用RSEM 计算基因的表达水平,表达量用FPKM表示。对比分析水杨酸喷施前后地黄块根中基因的表达水平,获取水杨酸喷施处理的特异响应基因,并对其进行GO注释和KEGG注释,根据注释结果进行功能分类和KEGG pathway分类,并使用R软件中的phyper函数进行富集分析,获得水杨酸喷施处理下地黄块根内的关键分子响应进程。
1.4 实时荧光定量(qRT-PCR)分析
用TaKaRa反转录试剂盒对RNA进行反转录,合成cDNA,反应体系包括1 μL oligo dT primer,1 μL dNTP Mixture,2 μg 模板RNA,加水补足体积到10 μL,65℃保温5 min后,冰上迅速冷却。再加0.5 μL的RNase抑制剂,1 μL PrimeScript Ⅱ RTase,4 μL 5 × PrimeScript Ⅱ Buffer,加水补足体积到20 μL。反应程序为42℃ 60 min,95℃ 5 min。以RgTIP41为内参基因,用实时荧光定量PCR检测基因表达水平。所用试剂盒为SYBR® Premix Ex Taq™ Ⅱ(Tli RNaseH Plus) (Takara,大连),使用仪器为Bio-Rad IQ5(上海伯乐公司)。定量反应体系为25 μL,包含2 μL 上述反转录cDNA产物,上、下游引物各1 μL,12.5 μL SYBR® Premix Ex Taq,8.5 μL ddH2O。反应程序为:95℃变性30 s;然后95℃ 5 s, 60℃ 30 s,40个循环。结束反应后获得不同样品的扩增循环数Ct,使用2-ΔΔCt法计算不同基因的相对表达量。
2. 结果与分析
2.1 SA处理对地黄毛蕊花糖苷含量的影响
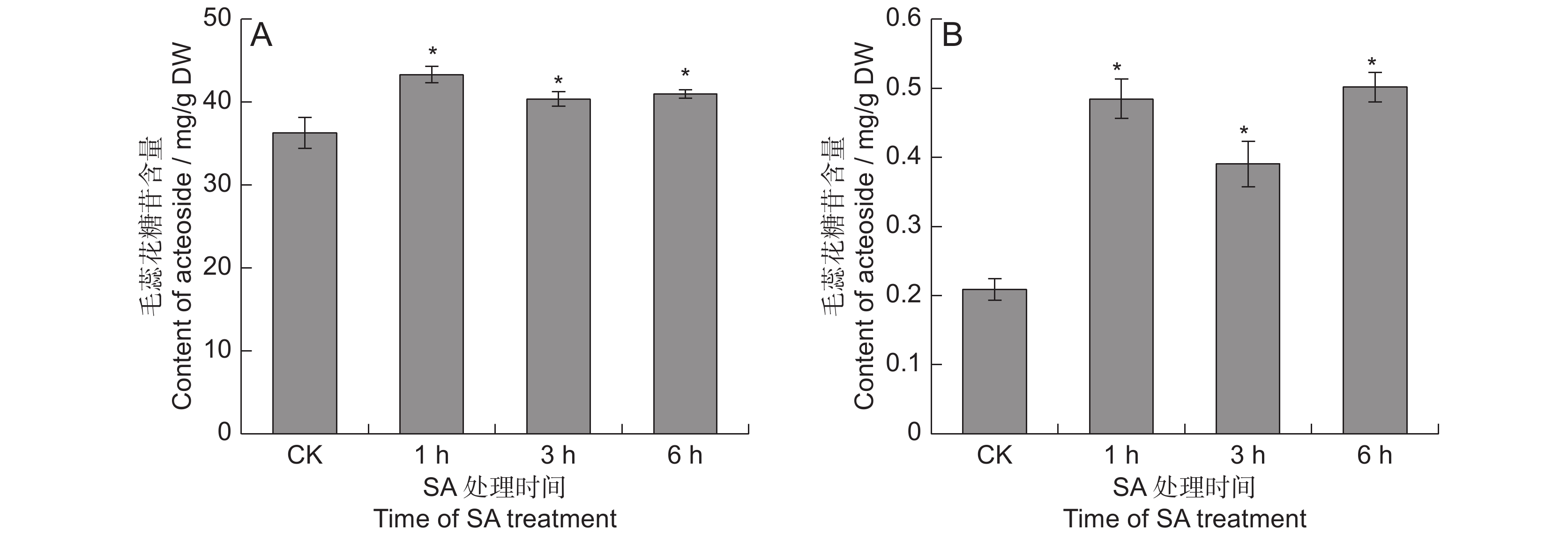
对SA处理的地黄叶和块根中的毛蕊花糖苷含量进行测定,结果表明,SA能够显著提高毛蕊花糖苷的含量(图1)。在叶中, SA处理1 ~ 6 h后,毛蕊花糖苷含量分别提高了11.2% ~ 19.3%。块根中毛蕊花糖苷的提升幅度远高于叶片,分别较对照提高了0.9 ~ 1.4倍,处理 6 h后的毛蕊花糖苷含量最高,达0.5 mg/g,远超2015版《中国药典》规定的0.02%,说明地黄叶面喷施SA可显著提高叶和块根中毛蕊花糖苷的含量。
![]() 图 1 SA处理下地黄叶和块根中的毛蕊花糖苷含量A:叶中毛蕊花糖苷含量;B:块根中毛蕊花糖苷含量。* 表示处理与对照差异显著,P < 0.05。Figure 1. Acteoside content in SA-treated leaves and tuberous roots of Rehmannia glutinosaA: Acteoside content in leaves; B: Acteoside content in tuberous roots. * indicates significant difference at P < 0.05 level.
图 1 SA处理下地黄叶和块根中的毛蕊花糖苷含量A:叶中毛蕊花糖苷含量;B:块根中毛蕊花糖苷含量。* 表示处理与对照差异显著,P < 0.05。Figure 1. Acteoside content in SA-treated leaves and tuberous roots of Rehmannia glutinosaA: Acteoside content in leaves; B: Acteoside content in tuberous roots. * indicates significant difference at P < 0.05 level.2.2 地黄块根RNA测序分析
利用Agilent 2100 Bioanalyzer和Fragment Analyzer分别对提取的各样品总RNA质量进行检测,结果显示,12个样品的总RNA浓度在430 ~ 1260 ng/μL,总RNA质量在8.6 ~ 25.2 μg,RNA的浓度和总量满足建库需求。RIN值在8.2 ~ 9.9,28S/18S > 1.6,说明RNA较为完整,符合建库要求。
为了分析SA处理后地黄块根相关基因的表达特性,对SA处理1、3和6 h后的地黄块根进行RNA-seq分析,结果表明(表1),每个测序样本获得的总原始读段量均为21.94 M,去除低质量、接头污染及未知碱基N含量过高的reads,获得的高质量reads在20.98 ~ 21.27 M,碱基数均在1.05 ~ 1.06 Gb,测序数据量基本一致。将每个样品的测序数据匹配地黄参考转录组,发现匹配率在85.04% ~ 87.70%,特异匹配率在47.35% ~ 51.31%,测序数据能够较好地反映细胞中基因表达的真实情况,说明测序质量良好,可以进行后续基因表达分析。
表 1 测序数据统计结果Table 1. Statistics of sequenced data样本
Sample总原始序列
Total raw reads / M总测序序列
Total clean reads / M总测序碱基数
Total clean bases / Gb测序序列比率
Clean read ratio / %总匹配率
Total mapped / %特异匹配率
Uniquely mapped / %Control_1 21.94 21.13 1.06 96.3 87.70 51.31 Control_2 21.94 21.07 1.05 96.04 86.92 51.18 Control_3 21.94 21.08 1.05 96.08 85.72 51.09 SA 1h_1 21.94 21.27 1.06 96.93 85.44 47.35 SA 1h_2 21.94 21.08 1.05 96.05 87.07 48.03 SA 1h_3 21.94 21.15 1.06 96.37 87.28 49.26 SA 3h_1 21.94 20.98 1.05 95.59 86.85 50.36 SA 3h_2 21.94 21.05 1.05 95.93 85.74 49.92 SA 3h_3 21.94 21.05 1.05 95.95 86.81 50.47 SA 6h_1 21.94 21.09 1.05 96.11 86.50 50.25 SA 6h_2 21.94 21.06 1.05 96.00 85.04 49.42 SA6h_3 21.94 21.03 1.05 95.86 86.68 49.97 2.3 SA处理前后差异表达基因分析及筛选
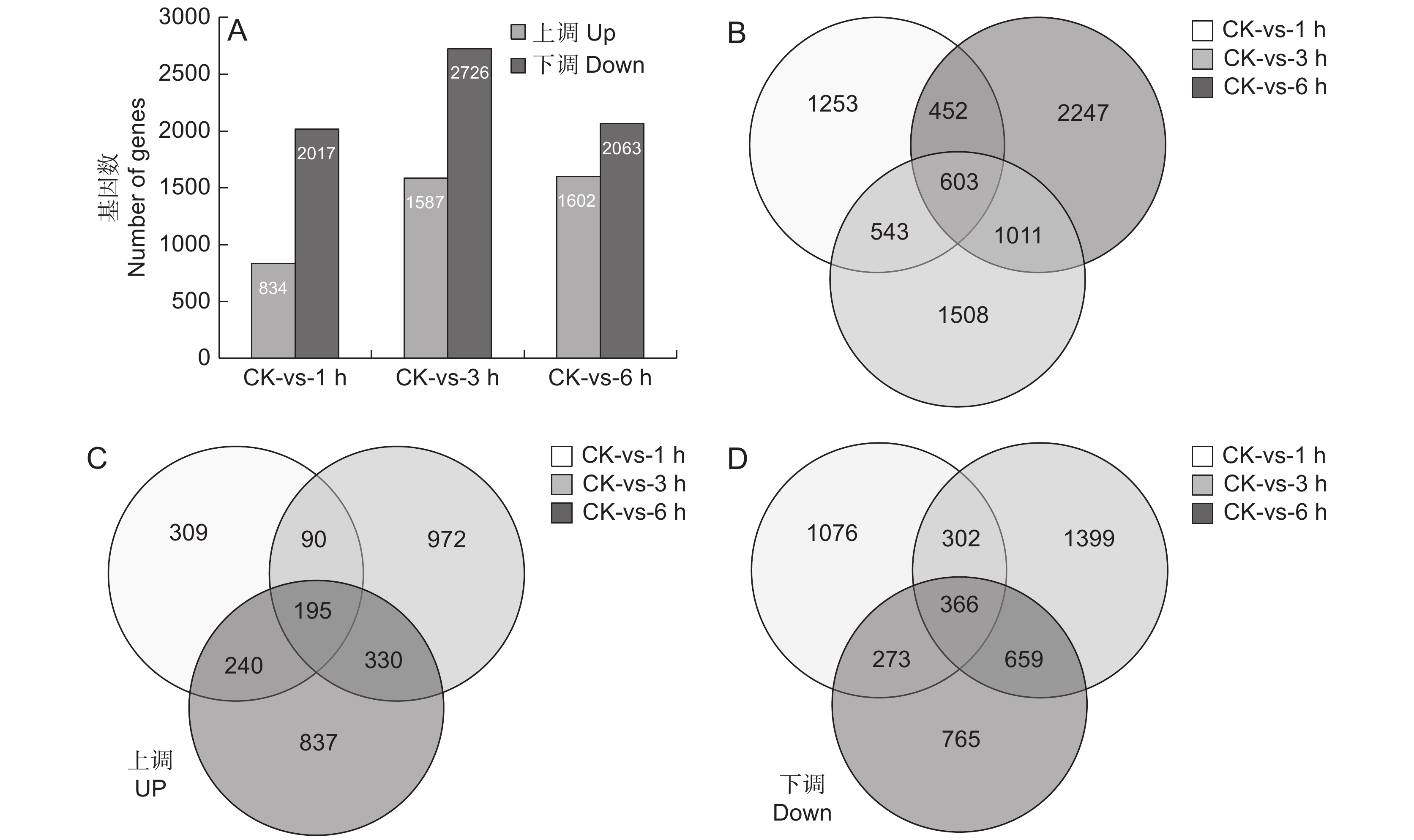
为了分析SA处理后块根中基因的表达特性,采用FPKM值比较基因丰富度的差异(图2)。使用以下标准对差异表达基因(DEGs)进行识别和筛选:校正P值 < 0.001且log2值 ≥ 2。分析SA处理不同时间后与对照样品中的差异表达基因(图2:A),发现SA处理1 h后834个基因上调,2017个基因下调;处理3 h后1587个基因上调,2726个基因下调;处理6 h后1602个基因上调,2063个基因下调。进一步分析SA处理后的3个时间点与CK相比的共同差异表达基因,发现共有603个基因是共同差异表达的(图2:B),其中上调表达和下调表达的基因数分别为195(图2:C)和366个(图2:D)。
![]() 图 2 SA处理后基因显著差异表达A:SA处理过程中上调和下调的基因数目; B ~ D:SA处理后不同时间点鉴别出的总DGEs(B)、上调DGEs(C)和下调DGEs的维恩图。Figure 2. Significant DEGs in response to SA treatmentA: Up-regulated and down-regulated gene numbers during SA treatment; B − D: Venn diagram of total DEGs (B), up-regulated DEGs (C), and down-regulated DEGs (D) identified at different time points after SA treatment.
图 2 SA处理后基因显著差异表达A:SA处理过程中上调和下调的基因数目; B ~ D:SA处理后不同时间点鉴别出的总DGEs(B)、上调DGEs(C)和下调DGEs的维恩图。Figure 2. Significant DEGs in response to SA treatmentA: Up-regulated and down-regulated gene numbers during SA treatment; B − D: Venn diagram of total DEGs (B), up-regulated DEGs (C), and down-regulated DEGs (D) identified at different time points after SA treatment.对603个DGEs进行GO功能分类(图3:A),结果显示其共分为分子功能(Molecular function)、细胞功能(Cellar function)和生物功能(Biological function)3个大类。其中分子功能分类中的催化活性(Catalytic activity)占比最多,其次是ATP结合;在细胞功能分类中,细胞(Cell)、细胞膜(Membrane)、细胞膜构件(Membrane part)较多;生物功能分类中,细胞过程(Cellular process)和代谢过程(Metabolic process)所占比重最多。进一步对603个DEGs进行KEGG代谢通路富集分析,图3:B展示了最显著的前20个代谢通路,这些通路涉及各项生命活动。其中首先被富集的是苯乙醇苷生物合成通路(Phenylpropanoid biosynthesis),其次是淀粉和蔗糖代谢合成通路(Starch and sucrose metabolism)、植物MARK信号通路(MAPK signaling pathway plant)及RNA聚合酶通路(RNA polymerase)。这说明SA喷施对地黄块根中次生代谢物的积累产生了较大影响,且调控了碳水化合物和MAPK等多个代谢通路。
![]() 图 3 SA处理后3种比对均差异表达基因的GO分类及KEGG通路富集A:603个基因的GO分类;B:KEGG富集的前20个代谢通路。圆点大小和颜色分别表示通路中DEGs的数量和Q值范围。1:分子转导活性;2:分子载体活性;3:信号转导活性;4:结构分子活性;5:转运活性;6:转录调节活性;7:抗氧化活性;8:结合;9:分子功能调节;10:催化活性;11:膜封闭腔;12:超分子复合物;13:共质体;14:细胞连接;15:细胞组分;16:细胞外区域;17:膜组分;18:膜;19:细胞器组分;20:细胞器;21:大分子复合物;22:细胞;23:生殖过程;24:繁殖;25:解毒作用;26:多细胞生物过程;27:发育过程;28:多生物体过程;29:信号;30:定域化;31:生物调节;32:对刺激的反应;33:代谢过程;34:细胞过程;35:细胞成分组织或生物合成。Figure 3. GO classification and KEGG pathway enrichment of co-DEGs in three comparisonsA: GO classification of 603 genes; B: Top 20 enriched KEGG pathways among 603 genes. Size and color of dot represent number and scope of DEGs in pathway, respectively. 1: Molecular transducer activity; 2: Molecular carrier activity; 3: Signal transducer activity; 4: Structural molecule activity; 5: Transporter activity; 6: Transcription regulator activity; 7: Antioxidant activity; 8: Binding; 9: Molecular function regulator; 10: Catalytic activity; 11: Membrane-enclosed lumen; 12: Supramolecular complex; 13: Symplast; 14: Cell junction; 15: Cell part; 16: Extracellular region; 17: Membrane part; 18: Membrane; 19: Organelle part; 20: Organelle; 21: Macromolecular complex; 22: Cell; 23: Reproductive process; 24: Reproduction; 25: Detoxification; 26: Multicellular organismal process; 27: Developmental process; 28: Multi-organism process; 29: Signaling; 30: Localization; 31: Biological regulation; 32: Response to stimulus; 33: Metabolic process; 34: Cellular process; 35: Cellular component organization or biogenesis.
图 3 SA处理后3种比对均差异表达基因的GO分类及KEGG通路富集A:603个基因的GO分类;B:KEGG富集的前20个代谢通路。圆点大小和颜色分别表示通路中DEGs的数量和Q值范围。1:分子转导活性;2:分子载体活性;3:信号转导活性;4:结构分子活性;5:转运活性;6:转录调节活性;7:抗氧化活性;8:结合;9:分子功能调节;10:催化活性;11:膜封闭腔;12:超分子复合物;13:共质体;14:细胞连接;15:细胞组分;16:细胞外区域;17:膜组分;18:膜;19:细胞器组分;20:细胞器;21:大分子复合物;22:细胞;23:生殖过程;24:繁殖;25:解毒作用;26:多细胞生物过程;27:发育过程;28:多生物体过程;29:信号;30:定域化;31:生物调节;32:对刺激的反应;33:代谢过程;34:细胞过程;35:细胞成分组织或生物合成。Figure 3. GO classification and KEGG pathway enrichment of co-DEGs in three comparisonsA: GO classification of 603 genes; B: Top 20 enriched KEGG pathways among 603 genes. Size and color of dot represent number and scope of DEGs in pathway, respectively. 1: Molecular transducer activity; 2: Molecular carrier activity; 3: Signal transducer activity; 4: Structural molecule activity; 5: Transporter activity; 6: Transcription regulator activity; 7: Antioxidant activity; 8: Binding; 9: Molecular function regulator; 10: Catalytic activity; 11: Membrane-enclosed lumen; 12: Supramolecular complex; 13: Symplast; 14: Cell junction; 15: Cell part; 16: Extracellular region; 17: Membrane part; 18: Membrane; 19: Organelle part; 20: Organelle; 21: Macromolecular complex; 22: Cell; 23: Reproductive process; 24: Reproduction; 25: Detoxification; 26: Multicellular organismal process; 27: Developmental process; 28: Multi-organism process; 29: Signaling; 30: Localization; 31: Biological regulation; 32: Response to stimulus; 33: Metabolic process; 34: Cellular process; 35: Cellular component organization or biogenesis.2.4 毛蕊花糖苷合成相关催化酶基因表达分析
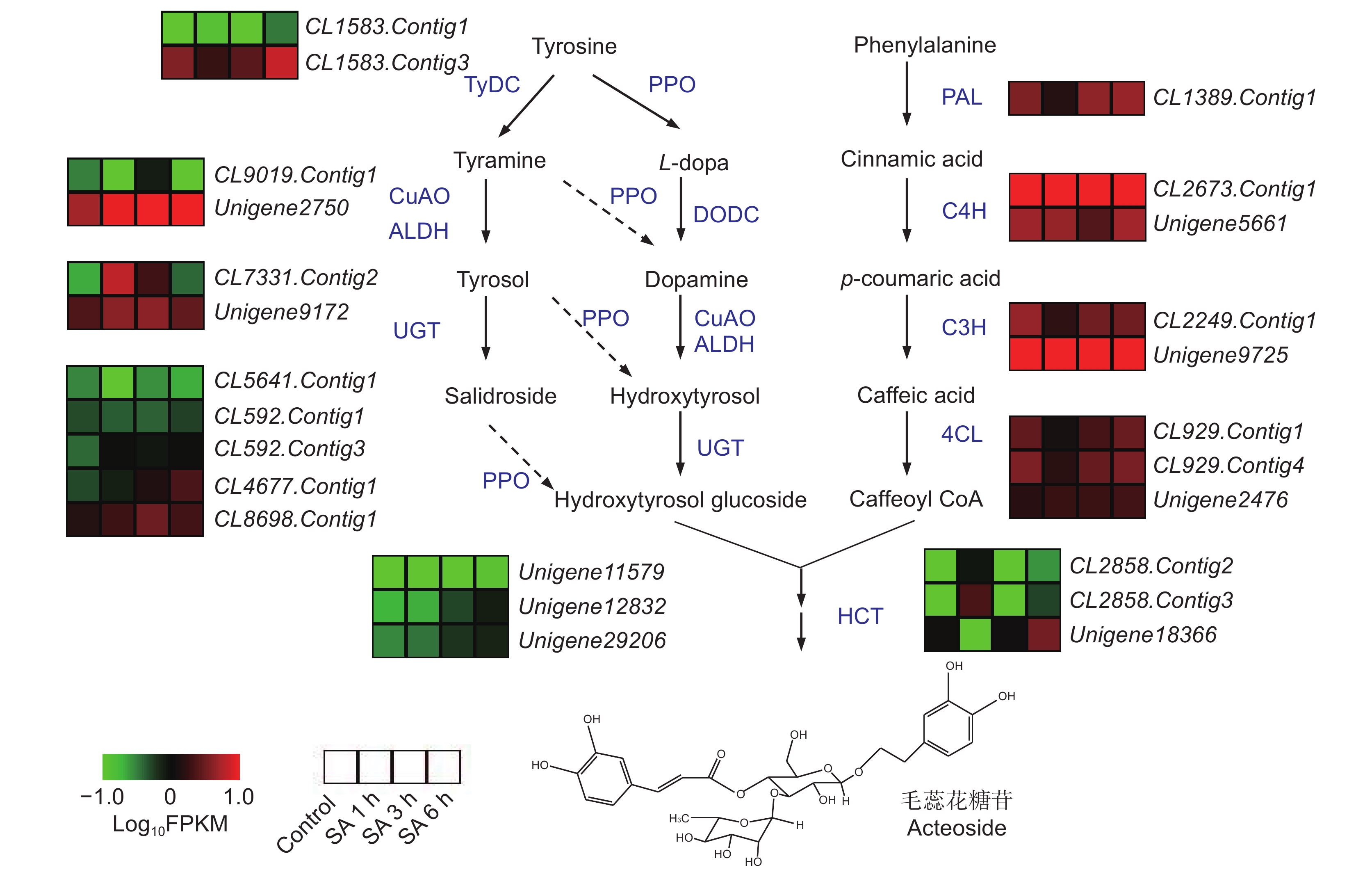
研究表明,在植物体内,毛蕊花糖苷由苯丙氨酸途径的咖啡酰辅酶A(Caffeoyl CoA)和酪氨酸途径的羟基酪醇苷(Hydroxtrosol glucoside)经缩合糖苷化后生成[8]。在地黄转录组中鉴定出可能参与毛蕊花糖苷合成的编码催化酶的基因215个,但SA处理后,地黄块根中仅有少数催化酶基因上调表达(图4)。在酪氨酸途径中,编码乙醛脱氢酶(ALDH)的基因CL7331.Contig2,在SA处理1 h和3 h后的地黄块根中表达量显著升高,另一个ALDH编码基因Unigene9172在SA处理后的表达量也有一定程度上升。编码糖苷转移酶(UGT)的基因CL4677.Contig1随着SA处理时间的延长其表达量逐渐升高,在SA处理6 h后表达量最高。编码多酚氧化酶(PPO)的两个基因Unigene12832和Unigene12832在SA处理3 h和6 h后的表达量增加较为明显。而苯丙氨酸途径的催化酶基因表达变化不明显。这说明SA处理后毛蕊花糖苷含量的增加可能主要与酪氨酸途径部分催化酶基因的表达量增加有关。
![]() 图 4 SA处理后毛蕊花糖苷合成相关的催化酶基因的表达Figure 4. Expression characteristics of enzyme genes involved in acteoside biosynthesis after SA treatment
图 4 SA处理后毛蕊花糖苷合成相关的催化酶基因的表达Figure 4. Expression characteristics of enzyme genes involved in acteoside biosynthesis after SA treatment2.5 差异表达转录因子筛选
对SA处理不同时间后地黄块根中差异表达的转录因子进行分析,结果表明(表2),25种转录因子的编码基因在处理前后呈差异表达。在处理1和3 h后,下调表达的转录因子较多,处理6 h后则上调表达的转录因子较多。WRKY、MYB、bHLH、AP2-EREBP、NAC和GRAS转录因子的差异表达基因较多,其中AP2-EREBP、WRKY和MYB的差异表达基因最多,且均表现为SA处理1 h后下调的基因较多, 3 h和6 h后上调表达的基因较多。进一步分析发现,共有20个转录因子编码基因在SA处理后的3个时间点均上调表达(表3),其中NAC和AP2-EREBP基因均为4个,WRKY基因有3个,MYB、GRAS、PLATZ基因各2个,bHLH、MADS和C2C2-CO-lik基因各1个。具有调控毛蕊花糖苷合成功能的RgWRKY37编码基因CL394.Contig2在SA处理1、3、6 h后的Log2(SA处理/CK)的值分别为0.79、0.82和0.57,Log2(SA处理/CK)的值虽然小于1,但其Q-value和P-value均达到了显著水平,说明在SA处理后CL394.Contig2上调表达。
表 2 SA处理后差异表达的转录因子数Table 2. Number of differentially expressed transcription factors (TFs) after SA treatment转录因子
Transcription factorCK-vs-1 h CK-vs-3 h CK-vs-6 h 共同差异表达的基因数
Number of common DEGs下调 Down 上调 Up 下调 Down 上调 Up 下调 Down 上调 Up zf-HD 2 1 1 1 0 0 0 WRKY 11 5 8 16 5 15 5 TUB 1 0 1 1 0 0 0 Trihelix 1 0 1 0 0 0 0 Tify 1 0 3 0 3 0 1 PLATZ 0 4 0 4 1 3 2 SRS 0 0 2 0 2 0 0 OFP 2 0 3 0 1 0 0 NAC 2 8 2 6 0 8 4 MYB 15 10 5 7 6 12 3 mTERF 1 0 2 0 1 1 0 MADS 2 2 3 4 2 2 1 LOB 1 0 4 2 2 0 0 HSF 5 2 4 6 3 6 2 GRAS 1 8 0 4 0 7 2 G2-like 2 0 2 1 0 3 0 CPP 2 0 2 0 4 0 2 C2H2 3 1 8 1 3 1 2 C2C2-GATA 2 0 1 0 5 0 0 C2C2-Dof 1 1 3 0 2 1 0 C2C2-CO-like 0 1 0 2 4 3 1 bZIP 1 2 0 0 1 0 0 bHLH 12 3 15 4 5 4 2 AP2-EREBP 15 12 12 18 9 21 6 ABI3VP1 5 2 7 1 1 1 0 总数 88 62 89 78 60 88 33 表 3 SA处理后上调表达的转录因子基因Table 3. Up-regulated transcription factor genes after SA treatment转录因子
Transcription factor基因
GeneLog2(SA/CK) 功能
Function1 h 3 h 6 h AP2-EREBP CL1637.Contig3 2.82 1.29 2.42 Ethylene-responsive transcription factor ERF071 AP2-EREBP CL4501.Contig2 1.66 1.75 2.16 Pathogenesis-related genes transcriptional activator PTI6 AP2-EREBP CL7827.Contig1 1.77 3.51 4.24 Ethylene-responsive transcription factor ERF106-like AP2-EREBP Unigene2558 1.62 1.06 1.74 Ethylene-responsive transcription factor 2-like MYB CL1983.Contig1 4.55 4.19 3.12 Transcription factor TFIIIB component B''-like MYB CL4303.Contig1 3.76 2.61 3.47 Single MYB histone protein NAC CL2945.Contig2 6.82 7.15 7.51 NAC domain-containing protein 82-like isoform X1 NAC CL4851.Contig1 3.15 1.99 2.44 NAC transcription factor 29 NAC CL4851.Contig2 3.15 2.27 2.82 NAC transcription factor 29 NAC Unigene5193 2.09 1.38 2.37 NAC domain-containing protein 72 C2C2-CO-like CL5505.Contig3 2.94 2.66 4.31 Zinc finger protein CONSTANS-LIKE 4-like PLATZ CL5569.Contig1 2.31 1.49 2.53 Interleukin-1 receptor-associated kinase 4 PLATZ CL5569.Contig3 2.47 1.71 2.38 Interleukin-1 receptor-associated kinase 4 GRAS CL645.Contig2 1.93 1.65 2.91 Scarecrow-like protein 14 GRAS Unigene10453 1.03 1.03 1.08 Scarecrow-like protein 15 WRKY CL6521.Contig1 2.84 1.66 2.35 Probable WRKY transcription factor 25 WRKY CL7324.Contig3 1.13 1.59 1.21 Probable WRKY transcription factor 35 WRKY CL791.Contig6 2.59 2.28 2.29 Probable WRKY transcription factor 40 bHLH Unigene12420 2.20 2.20 2.29 Phytochrome-interacting factor 3 MADS Unigene23315 2.17 3.01 2.27 MADS-box transcription factor 2.6 qRT-PCR验证基因表达差异
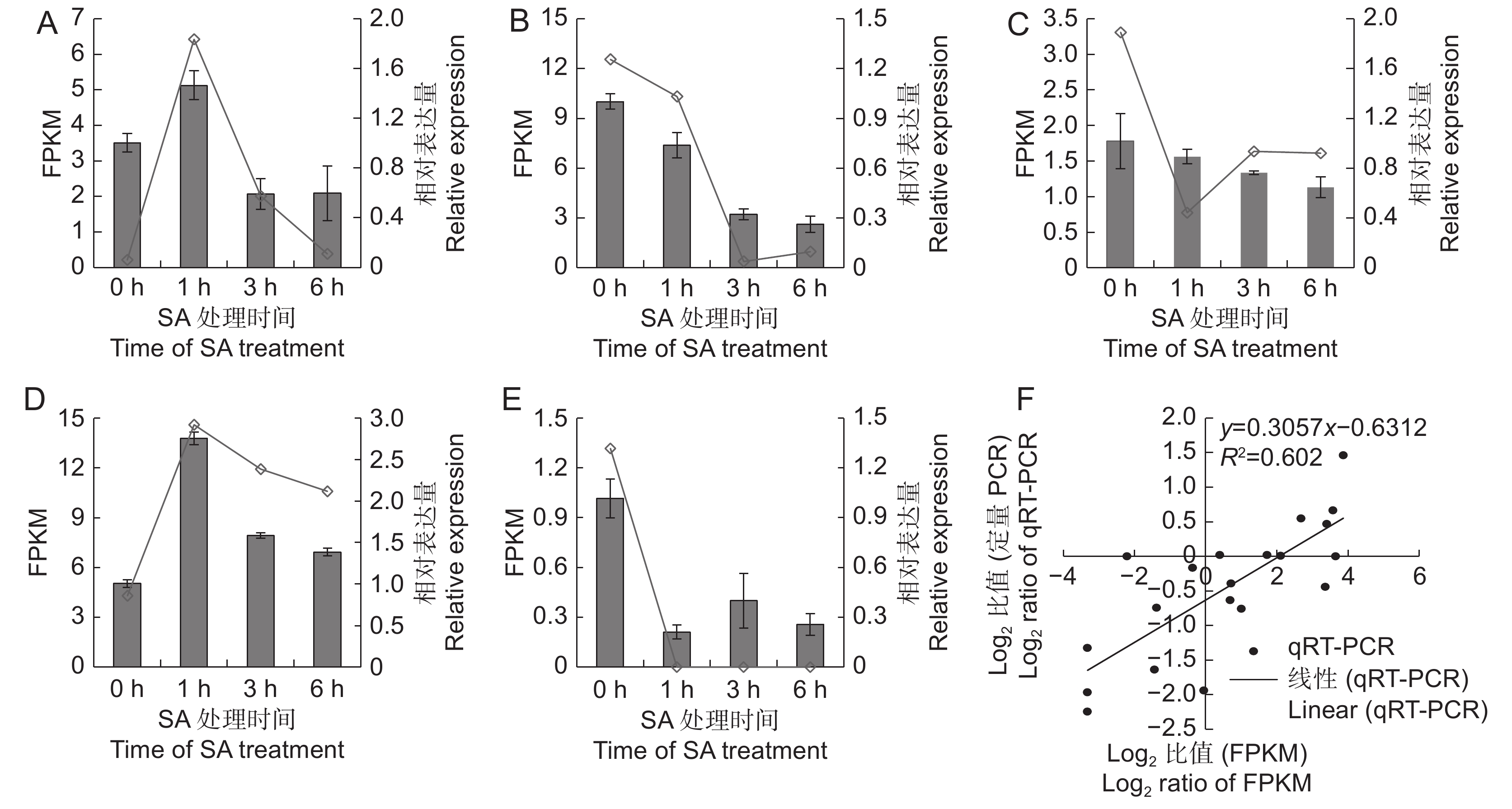
为了验证转录组测序对基因表达量分析的可靠性,随机选取CL7331.Contig2、Unigene1886、CL985.Contig1、CL5931.Contig2和CL379.Contig3等5个基因,利用qRT-PCR方法检测其在不同样本中的相对表达量。结果表明(图5),5个基因的定量结果与转录组获得的FPKM值变化趋势基本一致,其中CL7331.Contig2和CL5931.Contig2为SA处理后上调表达的基因,Unigene1886、CL985.Contig1和CL379.Contig3为SA处理后下调表达的基因。qRT-PCR与转录组测序的Pearson相关系数为0.602,相关性达显著水平,说明利用转录组测序分析SA处理后的基因表达量结果可靠。
![]() 图 5 差异表达基因的qRT-PCR验证A ~ E分别为CL7331.Contig2、Unigene1886、CL985.Contig1、CL5931.Contig2和CL379.Contig3的FPKM值与相对表达量;F:FPKM值与qRT-PCR的相关性分析。Figure 5. Validation of DEGs by qRT-PCRA–E: Represent expression and FPKM values of CL7331.Contig2, Unigene1886, CL985.Contig1, CL5931.Contig2, and CL379.Contig3; F: Correlation analysis between FPKM and qRT-PCR data.
图 5 差异表达基因的qRT-PCR验证A ~ E分别为CL7331.Contig2、Unigene1886、CL985.Contig1、CL5931.Contig2和CL379.Contig3的FPKM值与相对表达量;F:FPKM值与qRT-PCR的相关性分析。Figure 5. Validation of DEGs by qRT-PCRA–E: Represent expression and FPKM values of CL7331.Contig2, Unigene1886, CL985.Contig1, CL5931.Contig2, and CL379.Contig3; F: Correlation analysis between FPKM and qRT-PCR data.3. 讨论
植物次生代谢产物的积累既受自身遗传因素的控制,也受生长过程中生物与非生物环境条件的影响。一般而言,若药用植物的次生代谢产物在细胞中的含量相对较低,则会制约次生代谢产物的最终含量。近年来,人们常利用生物和非生物诱导子对植物进行处理,以提高植物特定次生代谢产物的生产[12]。如花生四烯酸(AA)、SA、茉莉酸甲酯(MeJA)和AgNO3均能够提高紫杉醇的含量[13]。50 µmol/L的乙烯利可以显著促进人参(Panax ginseng C. A. Meyer)根的生长和人参皂苷的积累[14]。诱导子提高苯乙醇苷含量的研究亦有报道,如外源添加Ag + 和腐胺均可以提高肉苁蓉(Cistanche deserticola Y. C. Ma)细胞培养物中松果菊苷和毛蕊花糖苷的含量[15]。本课题组前期研究发现,在地黄毛状根培养的培养基中添加25 μmol/L的SA可将毛蕊花糖苷的含量提高2.28倍[10]。本研究发现,叶面喷施100 μmol/L的SA可显著提高地黄叶片和块根中毛蕊花糖苷的含量,在块根中最高可提高1.4倍,说明在大田中叶面喷施诱导子可有效提高地黄块根中的次生代谢产物,有助于改善中药材的品质。
转录组测序分析不但可以高通量地获得基因表达的有关信息,还能够揭示基因表达与生命现象之间的内在联系,从而表征生命体的生理活动规律并确定其代谢特征[16]。目前,转录组测序不仅用于模式植物和大田作物生长发育及逆境胁迫响应关键基因的筛选[17-19],在药用植物次生代谢产物合成调控的结构基因和转录因子基因的挖掘中也有广泛应用[20-22]。由于地黄为同源四倍体物种,其基因组测序虽有报道[23],但作为参考基因组仍存在一些问题。因此,本研究利用课题组前期获得的地黄根、叶转录组为参考基因集进行分析,发现测序数据的特异匹配率偏低(50%左右),与地黄毛状根转录组测序的结果[10]类似,可能与其为同源四倍体物种有关。地黄叶片表面喷施SA后,上调表达的基因数少于下调表达,与SA处理的地黄毛状根结果[10]不同,这可能与本研究以大田地黄材料进行SA处理有关。本研究还发现,利用RNA-seq分析基因表达与qRT-PCR分析的结果相关系数仅为0.602,虽然达到显著相关,但未达到极显著相关水平,可能与地黄的基因组较大(约2.6 Gb),而采用RNA-seq测序获得的数据量较小有关。因此,对于基因组较大的物种,建议提高RNA-seq测序的深度,以获得更多的基因表达信息,提高基因表达量分析的准确性。
毛蕊花糖苷的生物合成途径目前已经比较清楚,其羟基酪醇基团来源于酪氨酸途径,咖啡酰基团来源于苯丙氨酸途径[24]。本课题组进一步推导、优化了毛蕊花糖苷的生物合成途径,认为毛蕊花糖苷是由羟基酪醇苷和咖啡酰辅酶A在莽草酸邻羟基肉桂酰转移酶(HCT)/毛蕊花糖苷合酶(AcS)和UGT的催化下合成[10]。周延清等[25] 基于地黄代谢组学分析获得了KEGG途径中的香豆酸-3-羟化酶(C3H),并克隆了其全长编码序列。李欣容等[26]根据SA处理下毛状根中催化酶基因的表达特性,鉴定并克隆了响应SA诱导的毛蕊花糖苷合酶基因RgAcS1。Yang等[27] 鉴定了4个酪氨酸脱羧酶(TyDC)基因,遗传转化发现过量表达RgTyDC2和RgTyDC4的地黄块根、纤维根、茎、嫩叶和成熟叶中的毛蕊花糖苷含量均显著高于野生型。Wang等[28]筛选了1个响应SA和H2O2诱导的WRKY转录因子基因RgWRKY37,功能研究发现RgWRKY37过量表达的毛状根转化体中毛蕊花糖苷和总苯乙醇苷的含量均显著高于对照。本研究发现,在SA处理的地黄块根中2个ALDH基因、1个UGT基因和2个PPO基因均上调表达,可能与块根中毛蕊花糖苷的含量增加有关。同时,在SA处理后的3个时间点,编码WKRY、NAC和AP2-EREBP等转录因子的20个基因均显著上调表达,其中RgWRKY37的表达量均明显增加。本研究结果为进一步探讨SA诱导毛蕊花糖苷合成的分子机理奠定了基础。
-
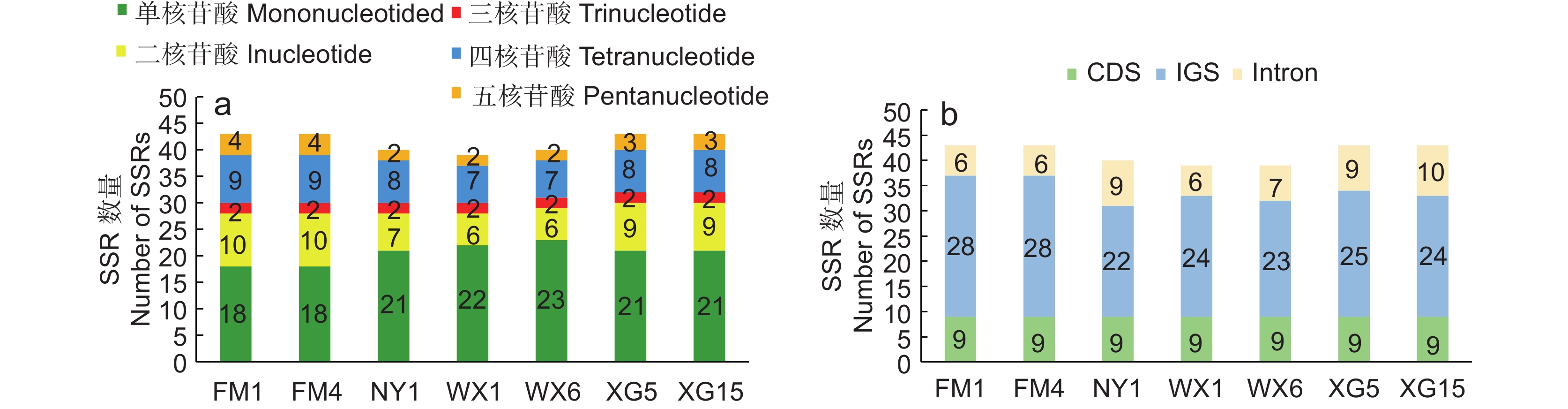
![]()
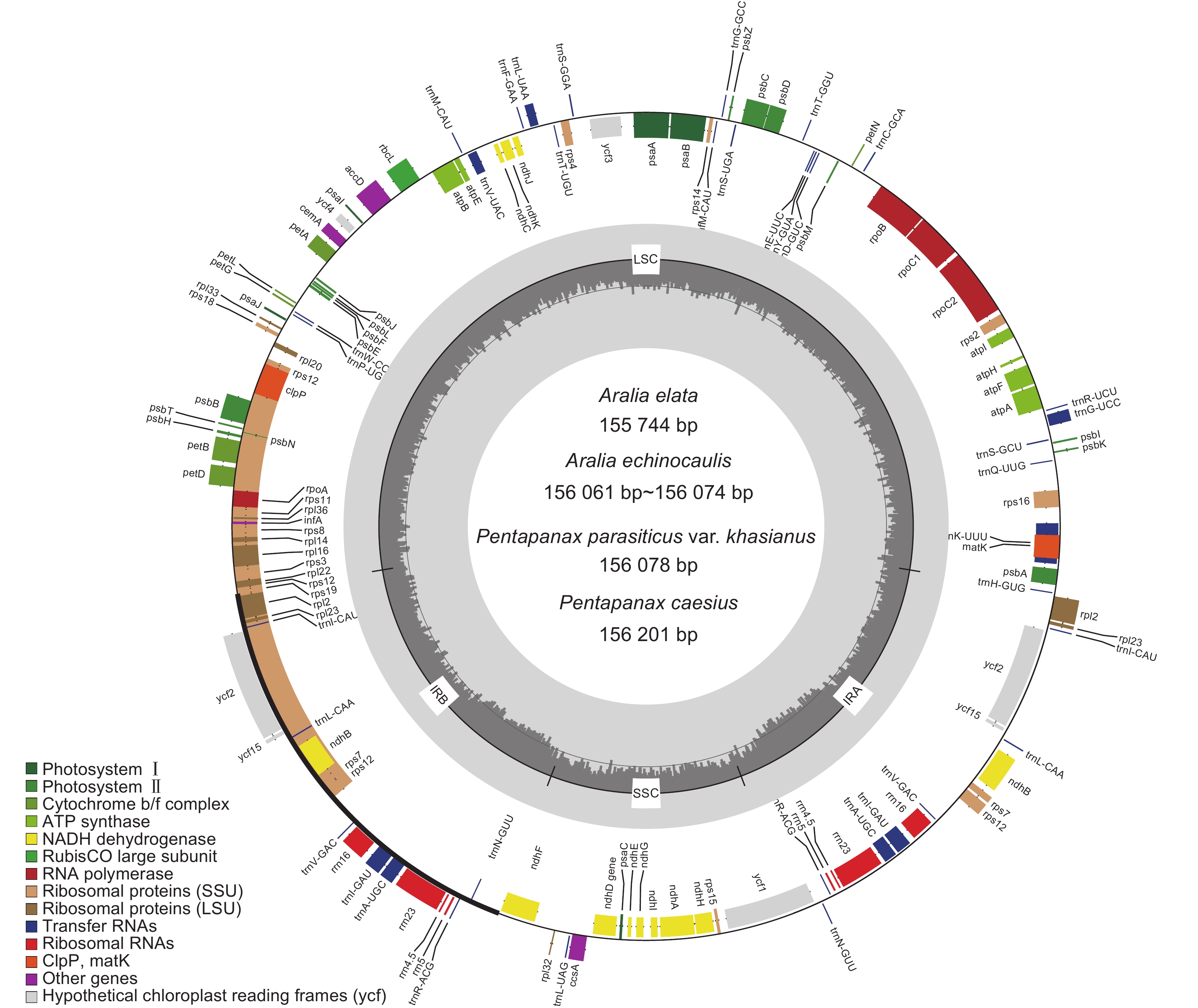
图 3 7个叶绿体基因组的SSR类型(a)及分布(b)
Figure 3. SSR type (a) and distribution (b) of seven chloroplast genomes
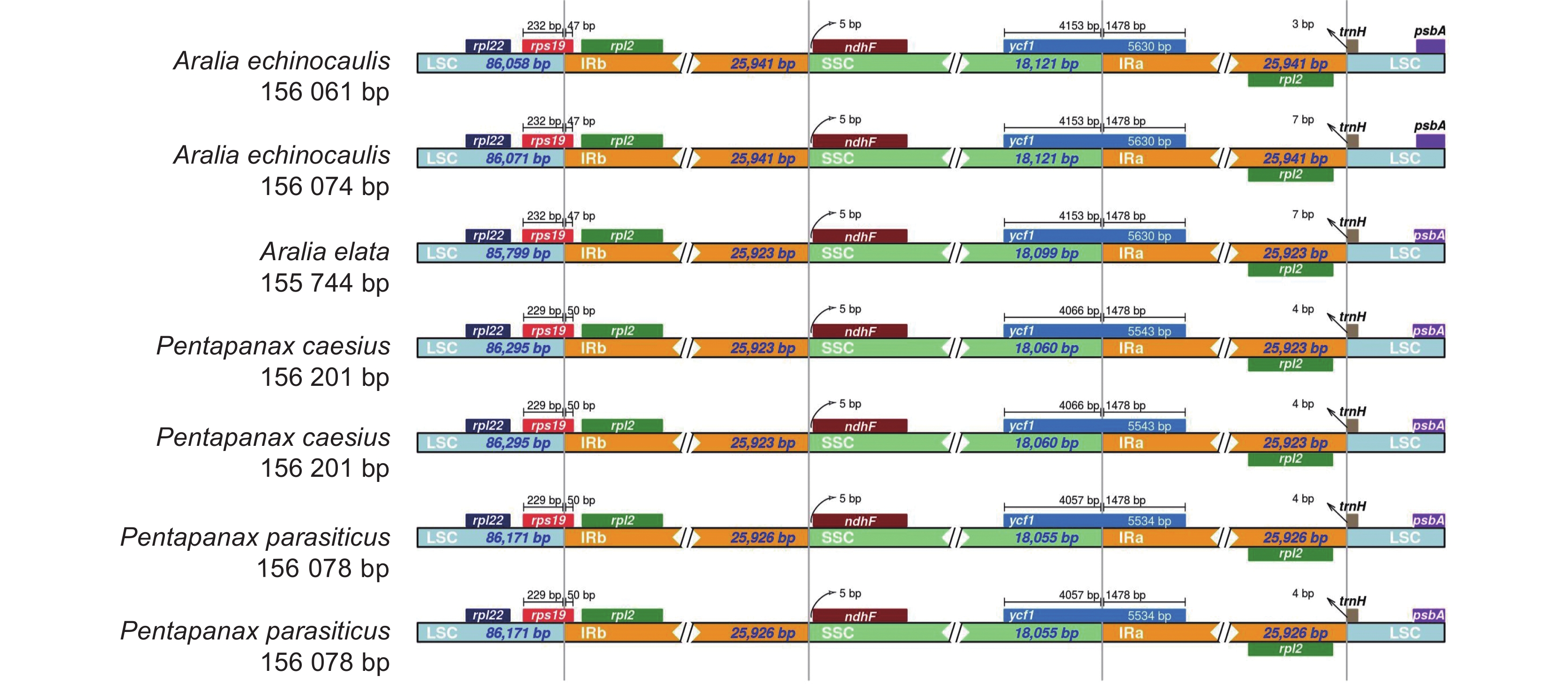
表 1 楤木属和羽叶参属物种采样信息
Table 1 Sampling information of Aralia and Pentapanax species
编号
No.中文名
Chinese name拉丁名
Latin name采集地
Collection places纬度
Latitude经度
LongitudeGenBank 登录号
GenBank No.FM1 毛梗寄生羽叶参 Pentapanax parasiticus var. khasianus
C. B. Clarke云南富民县 25°15'25"N 102°26'13"E ON493678 FM4 毛梗寄生羽叶参 P. parasiticus var. khasianus C. B. Clarke 云南富民县 25°15'25"N 102°26'13"E ON493679 NY1 楤木 Aralia elata (Miq.) Seem. 河南南阳 33°37'48"N 111°41'13"E ON493673 WX1 棘茎楤木 A. echinocaulis Hand. -Mazz. 云南维西 39°54'39"N 116°24'48"E ON493674 WX6 棘茎楤木 A. echinocaulis Hand. -Mazz. 云南维西 39°54'39"N 116°24'48"E ON493675 XG5 圆叶羽叶参 P. caesius (Handel-Mazzetti) C. B. Shang 云南香格里拉 27°50'87"N 99°42'26"E ON493676 XG15 圆叶羽叶参 P. caesius (Handel-Mazzetti) C. B. Shang 云南香格里拉 27°50'87"N 99°42'26"E ON493677  下载: 导出CSV
下载: 导出CSV
表 2 7个叶绿体基因组的基本特征
Table 2 Basic characteristics of seven chloroplast genomes
编号
No.长度Length / bp GC含量
GC / %基因数量 Gene number 基因组大小
Genome size大单拷贝区
LSC小单拷贝区
SSC反向重复区
IRrRNA tRNA CDS 总基因数
TotalFM1 156 078 86 171 18 055 25 926 38.10 37 8 87 132 FM4 156 078 86 171 18 055 25 926 38.10 37 8 87 132 NY1 155 744 85 799 18 099 25 923 38.10 37 8 87 132 WX1 156 074 86 071 18 121 25 941 38.10 37 8 87 132 WX5 156 061 86 058 18 121 25 941 38.10 37 8 87 132 XG5 156 201 86 295 18 060 25 923 38.10 37 8 87 132 XG15 156 201 86 295 18 060 25 923 38.10 37 8 87 132
下载: 导出CSV
表 3 7个叶绿体基因组的基因组成
Table 3 Gene composition of seven chloroplast genomes
基因分组 Gene group 基因名称 Gene name 核糖体大亚基 rpl2a ( × 2)、rpl14、rpl16a、rpl20、rpl22、rpl23 ( × 2)、rpl32、rpl33、rpl36 核糖体小亚基 rps2a、rps3、rps4、rps7 ( × 2)、rps8、rps11、rps12b ( × 2)、rps14、rps15、rps16a、rps18、rps19 RNA聚合酶 rpoA、rpoB、rpoC1a、rpoC2 rRNA基因 rrn4.5 ( × 2)、rrn5 ( × 2)、rrn16 ( × 2)、rrn23 ( × 2) tRNA基因 trnA-UGCa ( × 2)、trnC-GCA、trnD-GUC、trnE-UUC、trnF-GAA、trnfM-CAU ( × 2)、trnG-GCC、trnG-UCCa、trnH-GUG、trnI-CAU ( × 2)、trnI-GAUa、trnK-UUUa、trnL-CAA ( × 2)、trnL-UAAa、trnL-UAG、trnM-CAU、trnN-GUU ( × 2)、trnP-UGG、trnQ-UUG、trnR-ACG ( × 2)、trnR-UCU、trnS-GCU、 trnS-GGA、trnS-UGA、trnT-GGU、trnT-UGU、trnV-GAC ( × 2)、trnV-UACa、trnW-CCA、trnY-GUA 光系统Ⅰ psaA、psaB、psaC、psaI、psaJ 光系统Ⅱ psbA、psbB、psbC、psbD、psbE、psbF、psbH、psbI、psbJ、psbK、psbL、psbM、psbN、psbT、psbZ NADH脱氢酶 ndhAa、ndhBa ( × 2)、ndhC、ndhE、ndhF、ndhG、ndhH、ndhI、ndhJ、ndhK 细胞色素b/f 复合物 petA、petBa、petDa、petG、petL、petN ATP合成酶 atpA、atpB、atpE、atpFa、atpH、atpI 二磷酸核酮糖羧化酶大亚基 rbcL 成熟酶 matK 转录启动因子 infA 蛋白酶 clpPb 被膜蛋白 cemA 乙酰辅酶A羧化酶 accD 细胞色素C的合成基因 ccsA 保守假定的叶绿体开放阅读框 ycf1、ycf2 ( × 2)、ycf3b、ycf4、ycf15 ( × 2) 注:“a”表示基因含有1个内含子;“b”表示基因含有2个内含子;( × 2) 表明基因在IR 区复制。 Notes: “a” indicates that gene contains one intron; “b” indicates that gene contains two introns; ( × 2) indicates that gene was replicated in IR region.
下载: 导出CSV
-
[1] 程莹,李根有,夏国华,黄晌决,黄宇锋. 楤木属植物组织培养研究综述[J]. 浙江农林大学学报,2011,28(6):968−972. Cheng Y,Li GY,Xia GH,Huang SJ,Huang YF. Review on tissue culture of Aralia plants[J]. Journal of Zhejiang A & F University,2011,28 (6):968−972.
[2] 郑玲玲,裴凌鹏. 楤木属植物研究进展[J]. 中国民族医药杂志,2010,16(6):57−59. [3] 张广美,王春梅. 楤木属植物及其活性成分药理学研究进展[J]. 中华中医药学刊,2011,29(8):1715−1717. Zhang GM,Wang CM. Review of pharmacotogical effects of plants of Araliaceae and bioactive compounds[J]. Chinese Archives of Traditional Chinese Medicine,2011,29 (8):1715−1717.
[4] Wu ZY, Raven PH, Hong DY. Flora of China: Vol.13[M]. Beijing: Science Press & St. Louis: Missouri Botanical Garden Press, 2007: 480−489.
[5] 李湘萍,向其柏. 中国楤木属的研究[J]. 南京林业大学学报,1992,16(2):17−24. Li XP,Xiang QB. Studies on the genus Aralia Linn.[J]. Journal of Nanjing Forestry University,1992,16 (2):17−24.
[6] 向其柏. 五加科植物的新分类群及某些修订[J]. 南京林学院学报,1985,28(2):15−28. Xiang QB. New taxa and some revisions about the Araliaceae of China[J]. Journal of Nanjing Institute of Forestry,1985,28 (2):15−28.
[7] Wen J,Plunkett GM,Mitchell AD,Wagstaff SJ. The evolution of Araliaceae:a phylogenetic analysis based on ITS sequences of nuclear ribosomal DNA[J]. Syst Bot,2001,26 (1):144−167.
[8] Wen J. Revision of Aralia Sect. Pentapanax (Seem. ) J. Wen (Araliaceae)[M]. Fuzhou: Cathaya, 2002: 1−116.
[9] Wen J. Systematics and Biogeography of Aralia L. (Araliaceae): Revision of Aralia Sects. Aralia, Humiles, Nanae, and Sciadodendron[M]. Washington D. C: Contributions from the United States National Herbarium, 2011: 1−172.
[10] Li R,Wen J. Phylogeny and diversification of Chinese Araliaceae based on nuclear and plastid DNA sequence data[J]. J Syst Evol,2016,54 (4):453−467. doi: 10.1111/jse.12196
[11] Jansen RK,Raubeson LA,Boore JL,Depamphilis CW,Chumley TW,et al. Methods for obtaining and analyzing whole chloroplast genome sequences[J]. Methods Enzymol,2005,395:348−384.
[12] Alzahrani DA. Complete chloroplast genome of Abutilon fruticosum:genome structure,comparative and phylogenetic analysis[J]. Plants,2021,10 (2):270. doi: 10.3390/plants10020270
[13] 樊守金,郭秀秀. 植物叶绿体基因组研究及应用进展[J]. 山东师范大学学报(自然科学版),2022,37(1):22−31. Fan SJ,Guo XX. Advances in research and application of plant chloroplast genome[J]. Journal of Shandong Normal University (Natural Science)
,2022,37 (1):22−31. [14] Brown J,Pirrung M,McCue LA. FQC dashboard:integrates FastQC results into a web-based,interactive,and extensible FASTQ quality control tool[J]. Bioinformatics,2017,33 (19):3137−3139. doi: 10.1093/bioinformatics/btx373
[15] Jin JJ,Yu WB,Yang JB,Song Y,de Pamphilis CW,et al. GetOrganelle:a fast and versatile toolkit for accurate de novo assembly of organelle genomes[J]. Genome Biol,2020,21 (1):241. doi: 10.1186/s13059-020-02154-5
[16] Qu XJ,Moore MJ,Li DZ,Yi TS. PGA:a software package for rapid,accurate,and flexible batch annotation of plastomes[J]. Plant Methods,2019,15 (1):50. doi: 10.1186/s13007-019-0435-7
[17] Greiner S,Lehwark P,Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1:expanded toolkit for the graphical visualization of organellar genomes[J]. Nucleic Acids Res,2019,47 (W1):W59−W64. doi: 10.1093/nar/gkz238
[18] Amiryousefi A,Hyvönen J,Poczai P. IRscope:an online program to visualize the junction sites of chloroplast genomes[J]. Bioinformatics,2018,34 (17):3030−3031. doi: 10.1093/bioinformatics/bty220
[19] Beier S,Thiel T,Münch T,Scholz U,Mascher M. MISA-web:a web server for microsatellite prediction[J]. Bioinformatics,2017,33 (16):2583−2585. doi: 10.1093/bioinformatics/btx198
[20] Frazer KA,Pachter L,Poliakov A,Rubin EM,Dubchak I. VISTA:computational tools for comparative genomics[J]. Nucleic Acids Res,2004,32 (S2):W273−W279.
[21] Librado P,Rozas J. DnaSP v5:a software for comprehensive analysis of DNA polymorphism data[J]. Bioinformatics,2009,25 (11):1451−1452. doi: 10.1093/bioinformatics/btp187
[22] Katoh K,Misawa K,Kuma KI,Miyata T. MAFFT:a novel method for rapid multiple sequence alignment based on fast Fourier transform[J]. Nucleic Acids Res,2002,30 (14):3059−3066. doi: 10.1093/nar/gkf436
[23] Kalyaanamoorthy S,Minh BQ,Wong TKF,von Haeseler A,Jermiin LS. ModelFinder:fast model selection for accurate phylogenetic estimates[J]. Nat Methods,2017,14 (6):587−589. doi: 10.1038/nmeth.4285
[24] Zhang D,Gao FL,Jakovlić I,Zou H,Zhang J,et al. PhyloSuite:an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies[J]. Mol Ecol Resour,2020,20 (1):348−355. doi: 10.1111/1755-0998.13096
[25] Kim K,Nguyen VB,Dong JZ,Wang Y,Park JY,et al. Evolution of the Araliaceae family inferred from complete chloroplast genomes and 45S nrDNAs of 10 Panax-related species[J]. Sci Rep,2017,7 (1):4917. doi: 10.1038/s41598-017-05218-y
[26] Ran H,Liu YY,Wu C,Cao YN. Phylogenetic and comparative analyses of complete chloroplast genomes of Chinese Viburnum and Sambucus (Adoxaceae)[J]. Plants,2020,9 (9):1143. doi: 10.3390/plants9091143
[27] 田星,刘莹莹,张颖敏,杨从卫,钱子刚,李国栋. 藜芦属药用植物的叶绿体基因组比较分析和系统发育研究[J]. 中草药,2022,53(4):1127−1137. Tian X,Liu YY,Zhang YM,Yang CW,Qian ZG,Li GD. Comparative and phylogeny analysis of four Veratrum medicinal plants complete chloroplast genomes[J]. Chinese Traditional and Herbal Drugs,2022,53 (4):1127−1137.
[28] 黄琼林. 高良姜叶绿体基因组测序与特征分析[J]. 热带作物学报,2021,42(1):1−6. Huang QL. Complete sequencing and analysis of chloroplast genome from Alpinia officinarum Hance[J]. Chinese Journal of Tropical Crops,2021,42 (1):1−6.
[29] Liu J,Wen J. The complete chloroplast genome of Aralia atropurpurea (Araliaceae,the ginseng family) from the Sino-Himalayan region,China[J]. Mitochondrial DNA Part B Resour,2019,4 (2):2773−2774. doi: 10.1080/23802359.2019.1643805
[30] Kim CK,Kim YK. The complete chloroplast genome of Aralia cordata (Apiales:Araliaceae)[J]. Mitochondrial DNA Part B Resour,2019,4 (1):211−212. doi: 10.1080/23802359.2018.1546140
[31] Abdullah N,Henriquez CL,Mehmood F,Carlsen MM,Islam M,et al. Complete chloroplast genomes of Anthurium huixtlense and Pothos scandens (Pothoideae,Araceae):unique inverted repeat expansion and contraction affect rate of evolution[J]. J Mol Evol,2020,88 (7):562−574. doi: 10.1007/s00239-020-09958-w
[32] Liu CK,Yang ZY,Yang LF,Yang JB,Ji YH. The complete plastome of Panax stipuleanatus:comparative and phylogenetic analyses of the genus Panax (Araliaceae)[J]. Plant Divers,2018,40 (6):265−276. doi: 10.1016/j.pld.2018.11.001
[33] 宋菊,龙月红,林丽梅,尹峰,邢朝斌. 五加科植物叶绿体基因组结构与进化分析[J]. 中草药,2017,48(24):5070−5075. Song J,Long YH,Lin LM,Yin F,Xing ZB. Analysis on structure and phylogeny of chloroplast genomes in Araliaceae species[J]. Chinese Traditional and Herbal Drugs,2017,48 (24):5070−5075.
[34] Li R,Ma PF,Wen J,Yi TS. Complete sequencing of five Araliaceae chloroplast genomes and the phylogenetic implications[J]. PLoS One,2013,8 (10):e78568. doi: 10.1371/journal.pone.0078568
[35] Dong WP,Xu C,Li CH,Sun JH,Zuo YJ,et al. ycf1,the most promising plastid DNA barcode of land plants[J]. Sci Rep,2015,5:8348. doi: 10.1038/srep08348
[36] 郭栋梁,王静,韩冬梅,潘学文,李建光. 龙眼顶芽转录组简单重复序列(SSR)标记信息分析及分子标记开发[J]. 植物生理学报,2018,54(5):863−871. Guo DL,Wang J,Han DM,Pan XW,Li JG. Analysis on simple sequence repeat (SSR) information in apical transcriptome and development of molecular markers in Dimocarpus longan[J]. Plant Physiology Journal,2018,54 (5):863−871.
[37] 王久利,郑旭,邓因子. 暴马丁香的叶绿体基因组特征分析[J]. 阜阳师范大学学报 (自然科学版),2022,39(1):55−64. Wang JL,Zheng X,Deng YZ. Characterization of chloroplast genome of Syringa reticulata subsp. amurensis[J]. Journal of Fuyang Normal University (Natural Science)
,2022,39 (1):55−64. [38] 苏玥,刘娟娟,完斌,张鹏举,陈正根,等. 乳苣叶绿体基因组特征及其系统发育分析[J]. 中国农业科技导报,2021,23(6):33−42. Su Y,Liu JJ,Wan B,Zhang PJ,Chen ZG,et al. Chloroplast genome structure characteristic and phylogenetic analysis of Mulgedium tataricum[J]. Journal of Agricultural Science and Technology,2021,23 (6):33−42.
[39] 张韵洁,李德铢. 叶绿体系统发育基因组学的研究进展[J]. 植物分类与资源学报,2011,33(4):365−375. Zhang YJ,Li DZ. Advances in phylogenomics based on complete chloroplast genomes[J]. Plant Diversity and Resources,2011,33 (4):365−375.
[40] 许旭东,杨峻山,朱兆仪. 楤木属植物三萜皂甙研究进展[J]. 药学学报,1997,32(9):711−720. -
期刊类型引用(0)
其他类型引用(1)


计量
- 文章访问数: 231
- HTML全文浏览量: 52
- PDF下载量: 58
- 被引次数: 1
